AbbVie Receives U.S. FDA Approval of Once-Daily Viekira XR (dasabuvir, ombitasvir, paritaprevir and ritonavir) for the Treatment of Genotype 1 Chronic Hepatitis C
NORTH CHICAGO, Ill., July 25, 2016 /PRNewswire/ -- AbbVie (NYSE: ABBV), a global biopharmaceutical company, today announced that the U.S. Food and Drug Administration (FDA) has approved a New Drug Application (NDA) for Viekira XR (dasabuvir, ombitasvir, paritaprevir and ritonavir) extended-release tablets. Viekira XR is a once-daily, extended-release co-formulation of the active ingredients in Viekira Pak (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) and is for the treatment of patients with chronic genotype 1 (GT1) hepatitis C virus (HCV) infection, including those with compensated cirrhosis (Child-Pugh A). Viekira XR is not for people with decompensated cirrhosis.
Viekira XR is the first co-formulated three direct-acting antiviral (DAA) treatment for adult patients with GT1 HCV. Viekira XR is given once-daily as three oral tablets and must be taken with a meal. It is used without ribavirin (RBV) in GT1b patients and in combination with twice daily RBV in GT1a patients. The approval is supported by Phase 3 clinical trials for Viekira Pak which include data that demonstrated 100 percent sustained virologic response 12 weeks following treatment (SVR12) in GT1b patients with 12 weeks of therapy without ribavirin and 95 percent SVR12 in GT1a patients when used with ribavirin for 12 or 24 weeks of therapy.
"AbbVie's work continues to contribute to the transformation of hepatitis C care through our focus on evolving our current therapies as part of our ongoing commitment to patients," said Rob Scott, M.D., vice president, development and chief medical officer, AbbVie. "The approval of Viekira XR provides a new treatment option for genotype 1 hepatitis C patients in the U.S. with clinical trial data using the components of Viekira XR demonstrating 100 percent cure rates in genotype 1b patients."
There are six major HCV genotypes (GT1-6) and GT1 is the most prevalent form of HCV in the U.S., accounting for approximately 74 percent of all cases.1 Hepatitis C continues to be an important public health issue, with the Centers for Disease Control and Prevention (CDC) estimating that in the U.S. approximately 2.7 million people are chronically infected with HCV.2
The approval of Viekira XR is supported by data from seven Phase 3 clinical trials in more than 2,300 patients who received Viekira Pak with or without RBV for 12 or 24 weeks and two bioavailability studies comparing the formulations.
About Clinical Studies
The components of Viekira XR (administered twice daily with a meal) have been studied in seven Phase 3 clinical trials where 1076 subjects (including 181 with compensated cirrhosis) received the recommended regimen of Viekira +/? RBV for 12 weeks, or for 24 weeks in GT1a patients with compensated cirrhosis. Ninety-five to 100 percent achieved SVR12, which means the hepatitis C virus is not detectable in the blood three months after treatment ends. Cure rates varied by the subtype of hepatitis C and whether or not the person had cirrhosis. Individual results may vary.
USE
Viekira XR (dasabuvir, ombitasvir, paritaprevir, and ritonavir) extended-release tablets/Viekira Pak (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) (Viekira) are prescription medicines used with or without ribavirin to treat adults with genotype 1 chronic (lasting a long time) hepatitis C (hep C) virus infection.
Viekira can be used in people who have compensated cirrhosis.
Viekira is not for people with advanced cirrhosis (decompensated). If people have cirrhosis, they should talk to a doctor before taking Viekira.
About Viekira XR
The components of Viekira XR* have been studied in a broad range of genotype 1 (GT1) patients with chronic hepatitis C virus (HCV) infection, ranging from treatment-naïve to difficult to treat patients, such as those with compensated (mild, Child-Pugh A) cirrhosis of the liver, HCV/HIV-1 co-infection, liver transplant recipients with normal hepatic function and mild fibrosis, and those who have failed previous treatment with pegylated interferon (pegIFN) and ribavirin (RBV).
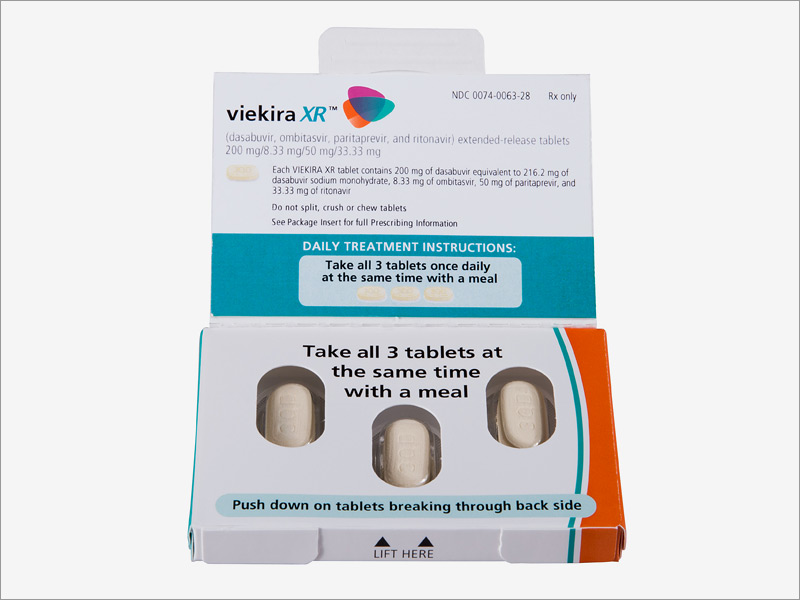
The extended-release co-formulation of these components, Viekira XR, consists of 200 mg of dasabuvir, 8.33 mg of ombitasvir, 50 mg of paritaprevir, and 33.33 mg of ritonavir per tablet, and is dosed three tablets once daily. Viekira XR must be taken with a meal, and tablets should be swallowed whole. People should not drink alcohol within four hours of taking Viekira XR. Viekira XR is contraindicated in patients with moderate to severe hepatic impairment (Child-Pugh B and C) due to risk of potential toxicity. Viekira XR is taken for 12 weeks, except in GT1a patients with cirrhosis and all liver transplant recipients with normal hepatic function and mild fibrosis, who should take it for 24 weeks. Ribavirin should be co-administered in GT1a patients and in all patients who have received a liver transplant.
Paritaprevir was discovered during the ongoing collaboration between AbbVie and Enanta Pharmaceuticals (NASDAQ: ENTA) for HCV protease inhibitors and regimens that include protease inhibitors. Paritaprevir is used in combination with AbbVie's ombitasvir with or without dasabuvir for the treatment of hepatitis C.
*Given as a fixed-dose combination of ombitasvir 25mg (an NS5A inhibitor), paritaprevir 150mg (an NS3/4A protease inhibitor), and ritonavir 100mg (an HIV-1 protease inhibitor), dosed once daily with a meal, and dasabuvir 250mg (a non-nucleoside NS5B palm polymerase inhibitor), dosed twice daily with a meal.
About AbbVie's Patient Assistance Program
AbbVie supports patient assistance programs to help qualified people access their needed AbbVie medication at no cost. In 2015, more than 81,000 U.S. patients received AbbVie's medicines at no cost3. For those who qualify, AbbVie plans to offer a patient assistance program for people taking Viekira XR. Since Viekira Pak's approval in 2014, AbbVie has supported access to medication for those living with chronic HCV and facing financial difficulties.
About AbbVie's HCV Clinical Development Program
AbbVie's HCV clinical development program is intended to advance scientific knowledge and the clinical care of people with chronic HCV infection. AbbVie is investigating a pan-genotypic (genotypes 1-6) regimen and is in Phase 3 of clinical development. For more information on AbbVie Phase 3 HCV studies, visit www.clinicaltrials.gov (NCT02243293).
About HCV
Hepatitis C is inflammation of the liver caused by an infection with the hepatitis C virus. It is transmitted when an infected person's blood enters the bloodstream of an uninfected person. The Centers for Disease Control (CDC) estimates that approximately 2.7 million people have chronic HCV infection in the U.S. There are six major HCV genotypes (GT1-6), with genotype 1 (GT1) as the most prevalent form in the U.S. It is estimated that of people infected with chronic HCV, about 5 to 20 percent will go on to develop cirrhosis over a period of 20–30 years, and with HCV-related liver transplants on the rise, HCV is a critical public health issue. Presently, there is no vaccine for HCV infection.
About AbbVie
AbbVie is a global, research-based biopharmaceutical company formed in 2013 following separation from Abbott Laboratories. The company's mission is to use its expertise, dedicated people and unique approach to innovation to develop and market advanced therapies that address some of the world's most complex and serious diseases. Together with its wholly-owned subsidiary, Pharmacyclics, AbbVie employs more than 28,000 people worldwide and markets medicines in more than 170 countries. For further information on the company and its people, portfolio and commitments, please visit www.abbvie.com. Follow @abbvie on Twitter or view careers on our Facebook or LinkedIn page.
Forward-Looking Statements
Some statements in this news release may be forward-looking statements for purposes of the Private Securities Litigation Reform Act of 1995. The words "believe," "expect," "anticipate," "project" and similar expressions, among others, generally identify forward-looking statements. AbbVie cautions that these forward-looking statements are subject to risks and uncertainties that may cause actual results to differ materially from those indicated in the forward-looking statements. Such risks and uncertainties include, but are not limited to, challenges to intellectual property, competition from other products, difficulties inherent in the research and development process, adverse litigation or government action, and changes to laws and regulations applicable to our industry.
Additional information about the economic, competitive, governmental, technological and other factors that may affect AbbVie's operations is set forth in Item 1A, "Risk Factors," of AbbVie's 2015 Annual Report on Form 10-K, which has been filed with the Securities and Exchange Commission. AbbVie undertakes no obligation to release publicly any revisions to forward-looking statements as a result of subsequent events or developments, except as required by law.
1 Wedemeyer H. Hepatitis C. Chapter 80: In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran's Gastrointestinal and Liver Disease. Vol 2. 10th ed. Philadelphia, PA: Saunders Elsevier; 2016.
2 Centers for Disease Control and Prevention (CDC). Hepatitis C FAQs for health professionals. http://www.cdc.gov/hepatitis/hcv/hcvfaq.htm#section1. Accessed June 9, 2016.
3 AbbVie 2016 Impact by the Numbers. http://www.abbvie.com/responsibility/home.html
SOURCE AbbVie































 Subscribe to us
Subscribe to us 100% Free.
100% Free.